L'industrie pharmaceutique fait partie des secteurs industriels soumis à un cadre réglementaire à la fois extensif par les domaines couverts et intensif par les degrés de liberté (ou de contraintes) qui lui sont imposés. Comme d'autres secteurs, ce cadre réglementaire est à la fois supranational et variable d'un pays à l'autre. Au niveau supranational, la protection de ses brevets fait l'objet de dispositions spécifiques dans le cadre de l'accord ADPIC (accord sur les droits de propriété intellectuelle qui touchent au commerce) ou TRIPS (Trade-Related Agreement on Trade-Related Property Rights) signé par les membres de l'OMC (Organisation mondiale du commerce) en 1994. Mais cet accord précise les conditions auxquelles un pays peut demander la levée de cette protection, pour produire un traitement à un prix sensiblement plus bas que celui pratiqué par l'entreprise. Cela a été le cas pour les premiers traitements antirétroviraux du Sida. Le débat est à nouveau ouvert sur les vaccins anti-Covid, pour lesquels les ONG réclament l'abandon de cette protection, au titre d'un problème majeur de santé publique. L'OMS (Organisation mondiale de la santé, agence spécialisée des Nations unies) se donne pour mandat de s'assurer que les traitements médicamenteux dits «essentiels» soient accessibles dans tous les pays à un prix abordable. Un outil d'intervention important est le Medicines Patent Pool (MPP), crée en 2005 pour assurer l'accès dans les pays émergents aux nouveaux traitements du Sida, de l'hépatite C et de la tuberculose. L'OMS utilise pour se faire deux leviers : l'octroi de licences dites « volontaires » à des fabricants des différents pays pour fournir des copies génériques des médicaments encore sous protection brevetaire, négocier des accords avec Les Entreprises du médicament (LEEM) pour obtenir la fourniture de produits à un prix inférieur à celui pratiqué dans les pays riches. L'acteur essentiel de cette politique est le fonds Unitaid.
Un deuxième exemple de réglementation supranationale est celui du marché européen. Bien que l'organisation des systèmes de santé et de leur financement reste du domaine réservé de chaque pays membre, la création de l'Agence européenne du médicament (European Medicines Agency, EMA) en 1995 a ouvert ce qui est maintenant le principal point de délivrance des autorisations de mise sur le marché (AMM). Les AMM sont une autorisation de commercialisation des produits, mais ne garantissent pas leur prise en charge financière par les différents systèmes d'assurance santé des pays membres. Mais outre l'accès au marché, l'EMA a la mission de s'assurer que le LEEM respecte un cadre normatif, par exemple en matière de processus de production (les good manufacturing practices, GMP). Pour certains produits, il impose aux industriels des plans de gestion des risques et peut être à l'origine d'une modification des indications d'un traitement et même de son retrait.
Parallèlement, l'Union européenne (EU) garantit la libre circulation des produits de santé entre les pays européens. Elle est garante d'une égalité de traitement des entreprises par les gouvernements des différents pays, par exemple en termes de délais d'accès aux différents marchés remboursés. Les pays sont en effet soumis à la Directive de transparence qui, comme son nom l'indique, impose des règles de transparence administrative sur les processus d'évaluation et de fixation des prix. Mais les entreprises sont soumises aux lois et aux règlements européens en matière d'antitrust.
Ce cadrage supranational coexiste avec des dispositifs nationaux différents d'un pays à l'autre, en fonction de son niveau de développement économique et de son système de financement des dépenses de santé. Ces dispositifs ont deux fonctions principales : l'évaluation du bénéfice en santé apporté par les nouveaux traitements, sous contrainte de leur sécurité et la place de ces traitements dans l'arsenal thérapeutique disponible ; la fixation du prix des traitements et des conditions de leur prise en charge par les systèmes existants d'assurance en santé. Pour les entreprises dont l'objectif est de vendre au plus grand nombre de pays, ce sont donc autant de contextes différents à prendre en compte. Elles doivent donc élaborer une stratégie globale en termes d'objectifs de prix, ainsi qu'une séquence de lancement de leur produit. De fait, une fonction nouvelle est apparue au sein des entreprises du médicament, l'accès au marché (market access).
Dans cet article, on présentera les éléments clés des stratégies de prix des labos, pour maximiser les conditions d'accès aux marchés solvabilisés par des systèmes développés d'assurance en santé. On commencera par un rappel du modèle économique de l'industrie du médicament. On pourra alors identifier quels sont les paramètres principaux de la stratégie de prix des entreprises. Face à ces stratégies, on présentera quels sont les principaux modèles existants d'évaluation de ces prix en donnant une place première au modèle français.
Le modèle économique de la pharmacie
Le processus de recherche et développement (R&D)
L'industrie pharmaceutique se caractérise par sa forte intensité en R&D et par les spécificités de ce processus. Le R pour recherche renvoie au processus de veille scientifique et technologique, dont l'objectif est de repérer les découvertes réalisées au sein des institutions académiques et des laboratoires internes. Passer de la découverte, par exemple d'un nouveau signal cellulaire pouvant être à l'origine d'une maladie, à la mise au point d'un produit permettant de l'inhiber est de la responsabilité d'équipes de recherche, soit académiques, soit en partenariat avec l'industrie.
Commence alors la phase dite préclinique, première étape du D du développement, dont l'objectif est de tester in vitro et in vivo l'activité et les effets de la molécule choisie. Le passage à la phase préclinique requiert l'approbation des autorités de mise sur le marché, qui doivent s'assurer de la sécurité du produit, sur la base des tests réalisés. L'objectif de cette étape est d'optimiser le produit sélectionné et de procurer suffisamment d'éléments probants en termes de sécurité et d'efficacité. Ce n'est qu'à la suite de cette démonstration que le laboratoire peut soumettre aux autorités de mise sur le marché un plan de développement clinique, dont le but est d'aboutir à la commercialisation du produit.
Ce processus comporte trois phases d'essais dits cliniques. Les essais de phase I ont pour objectif une évaluation préliminaire de l'efficacité et de la sécurité du produit, aux doses sélectionnées à la suite de la phase préclinique. Ce sont des essais de petite taille (pas plus de quelques dizaines de personnes incluses), sur volontaires sains, sans comparateur.
Le succès de cette phase permet d'initier la phase II du développement clinique, dont l'objectif principal est de déterminer la posologie optimale du médicament (le meilleur rapport efficacité/risques). Ces essais peuvent déjà donner des éléments d'efficacité comparative, contre placebo ou médicament actif. Ces essais peuvent être multiples et ils incluent des personnes malades, dont le nombre dépend de la pathologie étudiée et de la puissance statistique nécessaire pour obtenir des résultats valides d'un point de vue statistique.
Si la phase II est un succès, la phase III est la phase clé dont l'objectif est de démontrer l'efficacité du produit dans une indication, et ce par rapport soit à un placebo, soit à un comparateur actif. Sauf exception, la méthodologie de référence est l'essai randomisé en double aveugle (ni le prescripteur ni le malade ne connaissent le produit administré dans l'essai). Selon les pathologies et la puissance nécessaire pour montrer un bénéfice par rapport au comparateur choisi, ces essais peuvent inclure quelques centaines à quelques dizaines de milliers de malades.
La phase d'AMM est l'étape suivante. En règle générale, les entreprises présentent d'abord leurs dossiers d'enregistrement à la Food and Drug Administration (FDA) dont c'est la responsabilité pour le marché américain, puis à l'EMA, qui délivre l'AMM pour les vingt-sept pays de l'UE, enfin, à l'agence japonaise (Pharmaceutical and Medical Devices Agency, PMDA), trois ensembles économiques représentant environ 80 % (en chiffre d'affaires) du marché mondial du médicament.
L'AMM apporte la garantie que le produit considéré présente ce que l'on appelle un rapport bénéfice/risque positif : les bienfaits pour le patient l'emportent sur les risques inhérents au développement d'un produit actif ; il peut donc être commercialisé. Les modalités de cette commercialisation sont variables d'un pays à un autre. S'il existe des produits en vente libre (des produits sans prescription médicale), la majorité des produits sont soumis à prescription médicale et surtout à une prise en charge financière par des organismes d'assurance.
L'industrie pharmaceutique se caractérise donc par un processus long et incertain de développement de ses produits. À titre indicatif, on rappelle dans l'insert suivant les durées estimées des essais de phase I, II et III. La durée indiquée inclut la phase de recrutement des patients, pas la durée du suivi de chaque patient de l'essai, ni le temps d'analyse des résultats.
À partir d'une enquête auprès de dix grands groupes pharmaceutiques, représentant 35 % des cinquante premières firmes en termes de ventes et de dépenses de R&D, Di Masi et al. (2016) ont calculé les durées moyennes de chaque phase, ainsi que les risques d'échec entre chaque phase. Le tableau infra présente les résultats en termes de durée en mois. Les données sont relatives à la période 1995-2007.
Durées moyennes des phases et des transitions entre phase
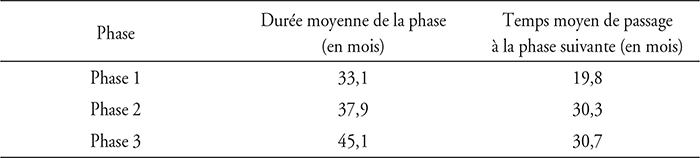
Source : Di Masi et al. (2016).
Il n'est pas possible d'additionner ces temps moyens qui sont calculés sur tous les projets dans une phase donnée et non de façon longitudinale. Mais ils permettent de donner un ordre de grandeur du temps requis pour amener un produit à l'enregistrement.
La durée de la procédure d'AMM dépend des règles adoptées par chaque agence. La durée du processus standard (standard review) de la FDA est fixée conventionnellement à dix mois. Cette durée est de 210 jours pour l'EMA. Ces durées n'incluent pas les phases d'interruption du processus d'analyse des données. En particulier, les agences peuvent arrêter le chronomètre (stopclock) pour demander aux promoteurs des produits des données complémentaires pour répondre aux interrogations à la suite d'une première évaluation. Enfin, la dernière étape est celle de l'accès aux différents marchés remboursés. La durée de ce processus est très variable en fonction des pays. En théorie, la directive européenne de transparence impose aux États membres une durée maximale de 180 jours après la soumission du dossier aux autorités, qui n'inclut pas les allers-retours entre les institutions engagées dans le remboursement et les laboratoires.
En résumé, en se fondant sur les données précédentes en ayant à l'esprit leurs limites, la durée moyenne des seules phases cliniques serait d'environ dix ans (115 mois), auxquelles il faudrait rajouter selon les cas entre dix et onze mois pour l'AMM et dans le meilleur des cas, en Europe, six mois pour l'accès à la prise en charge financière, soit en tout onze ans et demi, pour une durée de protection de la propriété intellectuelle de vingt ans.
Un deuxième facteur important est celui du pourcentage d'échec à chaque phase La publication la plus récente sur ce thème est celle de Pammolli et al. (2020), fondée sur une analyse de 50 000 projets entre 1990 et 2017. Sur la période 2010-2013, ils ont trouvé un taux de succès de la phase préclinique de 89,6 %, de 55,6 % pour la phase 1, de 80,5 % pour la phase 2 et de 68,8 % pour la phase 3. Le taux de succès cumulatif sur l'ensemble des phases est obtenu par la multiplication des taux intermédiaires, soit 27,5 %. Le taux de succès à l'enre gistrement étant de 28,7 %, ce sont donc au final 8 % des produits qui vont obtenir une AMM.
Le troisième facteur est financier. Le coût du développement d'un médicament fait l'objet de nombreuses publications et controverses, liées entre autres à l'accès aux données et aux méthodes de calcul adoptées. La mise sur le marché des vaccins contre le coronavirus a remis à l'ordre du jour les soutiens financiers publics à l'industrie pharmaceutique, qui améliore la rentabilité des fonds investis par les entreprises sans retour direct. Un autre débat est relatif à la prise en compte dans le calcul du coût de la mise sur le marché d'un produit du coût d'opportunité des dépenses investis. Ainsi, dans l'article le plus récent publié sur ce thème, Di Masi et al. (2016) ont réalisé une enquête auprès de dix entreprises multinationales et recueilli des données de dépenses de R&D. Le calcul des dépenses moyennes de R&D sans tenir compte du coût d'opportunité était de 1,395 Md$ et de 2,558 Md$ en tenant compte du coût du capital. Ce montant a été contesté au titre suivant : un biais de représentation de l'enquête, l'hypothèse étant faite que les coûts de R&D seraient plus importants dans les majors du secteur. Un deuxième argument tient aussi à la dispersion importante des résultats obtenus dans le calcul des coûts directs des différentes phases. Ainsi, Di Masi et al. (2016) ont estimé le coût moyen direct d'une phase I à 25,3 M$, avec un écart type de 29,6 M$, ce qui suggère une distribution des coûts très asymétrique. Les nombres respectifs pour les phases II et III étaient de 58,6 M$ (écart type 50,8) et 255,4 M$ (écart type 153,3).
On analyse à la section suivante l'impact de ces caractéristiques du processus de R&D sur les prix revendiqués par les entreprises du secteur. On peut cependant avancer les conclusions suivantes. Plus le coefficient d'attrition est important, plus les médicaments qui arrivent sur le marché doivent générer des ressources permettant de rémunérer leur propre coût de développement, le coût des échecs intervenus à chaque phase du processus, mais également les développements à venir. Plus le temps d'accès au marché est long, plus la durée d'exclusivité sera courte et donc le temps d'exclusivité du produit. Or, dans une aire de pathologie donnée, les laboratoires puisent tous dans le même réservoir de connaissances, ce qui implique une forme de mimétisme stratégique et une concurrence en termes d'accès au marché.
Les déterminants de la fixation du prix par l'entreprise
Les entreprises du secteur pharmaceutique présentent toutes les caractéristiques d'une situation monopolistique sur chaque produit nouveau qu'elles commercialisent. Cette affirmation doit être relativisée. Ce monopole dure le temps du brevet. Par ailleurs, un concurrent peut être très vite en position de proposer un produit sous brevet, mais qui cible la même pathologie ou le même groupe de patients. À ces réserves près, en situation de monopole pur, on dit que l'entreprise est price maker, et le client price taker. On verra cependant que ce n'est pas tout à fait le cas.
Comment l'entreprise construit-elle son offre de prix ? D'abord en fonction d'une rentabilité attendue de son produit, fonction des ventes espérées au niveau mondial. L'objectif de rentabilité est fixé par les principaux actionnaires de l'entreprise. Une stratégie mondiale de prix est élaborée, qui tient compte des différentes capacités à payer des grands marchés : le marché des États-Unis d'abord, puis le marché européen et le marché japonais. Il n'y a pas a priori de borne supérieure à ce prix, hors la question de son acceptabilité sociale, comme on a pu le voir récemment pour le prix des traitements de thérapie génique. Il s'agit d'un prix d'annonce, décliné par un grand marché et qui sert en outre à fixer des objectifs de prix final et de vente pour chaque filiale.
Ce prix n'est cependant pas décroché du prix des produits déjà existants et intègre la taille attendue du marché. À un extrême, le prix des traitements pour des maladies orphelines, qui comptent au niveau mondial quelques milliers ou dizaines de milliers de personnes malades, sera plus élevé « à valeur clinique égale » que pour un médicament pour l'hypertension, avec un marché mondial de plusieurs centaines de millions de patients. Le deuxième critère est celui du prix des comparateurs, médicaments déjà commercialisés pour la même cible.
Le prix intègre-t-il le coût du développement et de production du produit ? Oui, dans la mesure où il intègre le coût des échecs et son impact sur les ventres de l'entreprise. Non, dans la mesure où il n'existe pas de règle normative pour réaliser un partage équitable des dépenses directes et indirectes de ce développement. Il est de ce point de vue illusoire d'adopter une position tenue par les adversaires d'une industrie pharmaceutique capitaliste, consistant à fixer le prix d'un médicament sur la base de son prix de revient augmenté d'une marge considérée comme équitable. Le prix d'annonce est donc une variable stratégique qui marque la dimension monopolistique du secteur et notamment l'absence de transparence sur le niveau de prix demandé. Ce pouvoir monopolistique est par ailleurs accentué par l'espoir que suscitent certaines innovations thérapeutiques auquel il sera difficile pour l'acheteur de résister.
L'évolution récente de la R&D des grandes entreprises pharmaceutiques a accentué cet écart entre une logique industrielle de prix de revient pour fixer le prix d'un produit. En effet, la « big pharma » a connu au début du siècle une expiration brutale de nombreux brevets, ouvrant la porte à la commercialisation de médicaments dits géné riques, avec des baisses de prix pouvant atteindre selon les marchés de 50 % à 60 % de baisse. Cette baisse brutale de revenus s'est faite sur des produits dits de médecine de ville, pour des pathologies chroniques touchant des millions de personnes : maladies cardio-vasculaires, diabète, asthme, etc. Ces produits étaient issus d'un modèle de R&D éprouvé, où plusieurs laboratoires pouvaient trouver leur place en « imitant » les molécules de leurs concurrents : c'est l'ère des me-too, médicaments semblables dans leurs mécanismes d'action au premier entré sur le marché. Les laboratoires ont été plus ou moins habiles ou rapides à transformer leur fonction R&D, pour se tourner vers une nouvelle classe de médicaments issue des biotechnologies. Ce sont les centres de recherche académiques qui ont été les « découvreurs » de ces biotechnologies, certains se transformant en start-up, puis en entreprises dynamiques. L'américain Amgen a bâti son succès sur la mise sur le marché de l'érythropoïétine. Roche a misé très tôt sur un partenariat avec Genentech, pour produire notamment un traitement radicalement nouveau du cancer du sein, le trastuzumab, dont l'efficacité repose sur l'identification d'une mutation génétique. Les investissements des autres grands laboratoires ont été plus tardifs. Mais un effet de cette externalisation de la R&D a été de créer un marché très dynamique et pour certains spéculatifs de l'achat de start-up (ou d'accord de codéveloppement) par les grands laboratoires. Dans ce cas, la dépense à amortir est le prix de ces acquisitions. L'exemple récent qui a frayé la chronique est le rachat en 2011 de Pharmasset par Gilead, laboratoire américain, pour un montant d'environ 11 Md$ et un portefeuille de produits antiviraux. Le Sovaldi© est un traitement très efficace de l'hépatite C, dont le prix initial a créé une onde de choc et des débats passionnés sur l'écart entre le prix du comprimé (700 euros à l'époque) et son coût de production de 2,50 euros.
En résumé, les grands laboratoires pharmaceutiques établissent maintenant leur prix en fonction du coût d'acquisition des molécules et des caractéristiques du contrat d'achat ou de partenariat avec les entreprises qui ont découvert et réalisé les premiers développements d'une innovation.
La logique des « payeurs »
On utilise le terme « payeur » par commodité pour représenter les organisations ou les institutions qui interviennent dans l'accès au marché financé par des mécanismes d'assurance. Le « payeur » a deux fonctions : il doit estimer la valeur du produit pour la collectivité qu'il représente, il doit en fixer le prix et les conditions d'utilisation.
De façon schématique, il existe deux écoles d'estimation de la valeur d'un produit nouveau. La première école, que l'on illustrera plus loin par l'exemple français (et qui prévaut à quelques différences près en Allemagne), repose principalement sur l'analyse comparative des bénéfices cliniques d'un nouveau produit par rapport aux produits existants, pour la communauté desservie. C'est alors cette estimation qui va orienter la négociation entre l'industriel et le payeur, sur la base d'un principe simple. Si cet apport est considéré comme faible (voire inexistant), le prix du promoteur est contesté et la position de référence du payeur dans la négociation sera le prix des comparateurs. Si cette valeur est considérée comme importante, le payeur tentera néanmoins d'obtenir un prix plus bas que celui proposé par l'entreprise, mais ne sera plus en position de force. En effet, dans ce cas, ne pas aboutir à un accord peut conduire à ne pas mettre sur le marché un « bon » produit et entraîner une perte de chance pour les potentiels malades bénéficiaires.
La deuxième école, dont le Royaume-Uni est le porte-drapeau, est fondée sur une approche « utilitariste » de la valeur. La valeur clinique d'un produit et les conditions optimales de son utilisation du point de vue du rapport bénéfice/risque sont associées à une analyse économique, des études de coût-efficacité, qui introduit un coût d'opportunité de la dépense en santé (Le Pen et Lévy, 2019). Ce modèle fonctionne dans le cadre d'un budget de dépenses plafonné, dont il convient de maximiser le rendement en termes de quantité de santé produite. Les économistes de la santé anglais ont en effet imposé une mesure devenue quasi universelle de cette quantité de santé : c'est le QALY (quality adjusted life years, nombre d'années de vie en bonne santé). Dans un tel modèle, si le coût marginal d'une année de vie supplémentaire en bonne santé dépasse un certain seuil, le traitement en question ne doit pas être pris en charge par la collectivité. En effet, l'excès de dépenses au-delà du seuil pourrait être utilisé de façon plus efficiente, pourrait produire plus de santé.
Le cas français : un prix fondé sur la valeur clinique des médicaments
En France, le processus de valorisation des traitements et de fixation de leurs prix fait intervenir deux acteurs principaux. La Haute Autorité de santé (HAS) est une agence d'expertise scientifique dont l'indépendance est garantie par ses statuts. Elle est gouvernée par un collège dont les six membres sont nommés pour six ans par le gouvernement, avec un renouvellement par tiers tous les deux ans. Les « collégiens » sont inamovibles. Ce collège est doté d'un président, personnalité scientifique de haut niveau. Chaque membre du collège est de droit président d'une commission spécialisée, dont les plus importantes pour notre propos sont la Commission de la transparence (CT) qui évalue les médicaments et les vaccins et la Commission d'évaluation économique et de santé publique (CEESP), dont on détaillera le rôle ci-dessous. Le Comité économique des produits de santé (CEPS) est un comité interministériel sous la tutelle du ministère de la Santé et du ministère des Comptes publics. L'assurance maladie est représentée au sein du CEPS.
Schématiquement, la CT a pour mission de rendre des avis sur deux points importants : l'admission d'un traitement au remboursement de l'assurance maladie obligatoire, l'estimation de la valeur ajoutée d'un traitement par rapport aux traitements existants.
Un traitement sera remboursé s'il présente un Service médical rendu (SMR) satisfaisant. Pour établir ce SMR, la CT se fonde sur l'avis d'autorisation sur le marché de l'Agence européenne du médicament, mais va prendre en compte d'autres critères : la sévérité de l'affection et l'existence d'un bénéfice collectif attendu. La sévérité de l'affection sert principalement à moduler le ticket modérateur, qui sera octroyé in fine par l'assurance maladie. Dans la plupart des cas, le SMR sera déclaré important et l'assurance maladie remboursera le produit à hauteur de 65 %. Si le SMR est modéré, ce taux sera de 30 %, de 15 % s'il est faible et il peut être considéré comme insuffisant.
L'estimation de la valeur ajoutée d'un traitement est l'objet de l'établissement de l'amélioration du service rendu (ASMR). Il s'agit d'une cotation à cinq niveaux :
ASMR I, amélioration majeure ;
ASMR II, amélioration importante ;
ASMR III, amélioration modérée ;
ASMR IV, amélioration mineure ;
ASMR V, amélioration inexistante, signifie « absence de progrès thérapeutique ».
La CT donne également une estimation de la « taille de la population cible » : il s'agit du nombre de patients qui pourront potentiellement bénéficier du traitement. Cette population cible représente en quelque sorte la taille maximale du marché du produit.
L'évaluation de l'ASMR joue un rôle central dans la négociation de prix qui va s'engager ensuite avec le CEPS. En effet, depuis plusieurs années, les règles du jeu de cette négociation sont encadrées par une convention cadre, établie entre les pouvoirs publics représentés par le CEPS et l'organisme représentatif de l'industrie pharmaceutique en France, le LEEM. Cette convention est renégociée tous les quatre ans. De façon très schématique, cette convention stipule que si la CT a octroyé une ASMR de I à III, dite ASMR innovante, l'entreprise peut se voir garantir le prix qu'elle demande au lancement, avec une protection de ce prix sur une période de cinq ans. Plus exactement, le CEPS garantit que ce prix ne sera pas inférieur au prix le plus bas pratiqué sur les quatre principaux marchés européens (Allemagne, Italie, Espagne et Royaume-Uni).
Dans le cas d'une ASMR V, par définition le produit ne présente pas d'avantage par rapport aux produits existants et son prix devra être inférieur. Une ASMR IV va ouvrir une négociation sur la prime de prix que le payeur est prêt à accepter par rapport aux prix des comparateurs. Dans ce cas, le fait que le surcoût lié au nouveau traitement soit compensé par des économies liées à ses effets peut jouer un rôle. Il convient de préciser que cette présentation schématique ne rend pas compte d'un aspect important de la convention de prix qui sera signée à la fin de la négociation. Cette négociation aboutit en effet à ce que l'on appelle un prix facial (prix public officiel), mais la convention comportera généralement des clauses de remises qui peuvent prendre plusieurs formes. La forme principale est un accord prix-volume, l'entreprise s'engageant à reverser à l'assurance maladie un montant lié à un dépassement par rapport à un volume prévisionnel de ventes. Donc le prix final affiché en pharmacie après ajout de la marge des grossistes et des pharmaciens n'est pas le prix que paiera in fine l'assurance maladie.
À cette fixation initiale des prix s'ajoutent d'autres mécanismes de « gestion dynamique des prix ». Tous les ans, l'industrie pharmaceutique est tenue de contribuer à l'effort de maîtrise des dépenses de santé en consentant des baisses de prix. Enfin, l'évolution annuelle du chiffre d'affaires du secteur en matière de médicaments remboursables est fixée par la loi de financement de la Sécurité sociale (LFSS). Cette régulation puissante par les prix explique pourquoi la part des dépenses de médicaments de ville dans la consommation des biens et des services médicaux est passée d'environ 22 % en 2000 à 16 % en 2018, alors que les volumes de prescription n'ont cessé d'augmenter (de Pouvourville, 2021).
Le modèle anglais
En Angleterre et au Pays de Galles, l'accès à la prise en charge d'un médicament et son prix sont de la responsabilité de trois institutions : le ministère de la Santé (Department of Health), le Service national de santé (National Health Service, NHS) et une instance scientifique similaire à la HAS, le National Institute for Health Care and Excellence (NICE). Par comparaison avec la France, le NHS est une forme d'entreprise publique qui opère les services de santé.
En théorie, le prix du médicament est libre dans l'espace des deux royaumes. La décision stratégique ne porte donc pas sur ce paramètre, mais sur l'acceptation de la prise en charge d'un médicament au prix proposé par l'entreprise. Le NICE est le garant scientifique de cette décision, qui est bâtie sur une double évaluation : une évaluation clinique, proche dans son esprit de celle que réalise la CT en France, mais plus orientée sur une analyse et des recommandations en termes d'utilisation des produits sur le marché, et une analyse économique dont on a esquissé les principes. L'avis du NICE sur la pertinence de la prise en charge par le NHS d'un médicament ou d'un traitement repose sur sa « rentabilité » collective. Celle-ci est calculée comme étant le rapport entre les différences de coût de mise en œuvre d'un traitement et des traitements existants, et la quantité supplémentaire de santé apportée par le nouveau traitement. En anglais, ce ratio s'appelle l'ICER (incremental cost-effectivness ratio) ; l'équivalent français est le RDCR (ratio différentiel coût efficacité) (cf. schéma infra).
Représentation simplifiée du RDCR
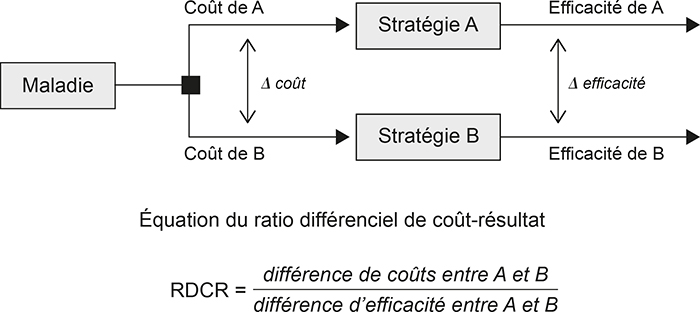
Source : Puyou de Pouvourville (2016).
L'utilisation des résultats de ce calcul pour un traitement nouveau, au prix proposé par le laboratoire, requiert deux paramètres importants. La logique du calcul implique le choix d'un seuil au-delà duquel le produit ne sera pas pris en charge : il faut fixer un seuil, qui représente le coût d'opportunité de la dépense publique en santé. Il faut disposer d'une métrique de l'efficacité des traitements, qui soit commune à tous les traitements et toutes les aires thérapeutiques.
Le choix d'un seuil fait l'objet de controverses théoriques et pratiques. Existe-t-il une « bonne » méthode pour choisir ce seuil pour les dépenses de santé ? La réponse est non. Il existe plusieurs méthodes, aboutissant à des valeurs différentes (HAS, 2014 ; Téhard et al., 2020). Pendant longtemps, les économistes ont raisonné par référence aux choix passés. Ainsi, les premières références à un seuil au-delà duquel un traitement ne serait pas coût-efficace ont été celle du ratio coût/efficacité de la dialyse, sur la base du raisonnement suivant : puisque la plupart des pays ont consenti à investir dans ce traitement, considéré comme très coûteux au moment de sa généralisation et permettant de sauver des vies, on peut consentir que des traitements avec un ratio coût-efficacité plus faible doivent être acceptés sans trop de discussions. En revanche, la prise en charge de traitements moins efficients pourrait se discuter. Ce raisonnement a conduit à utiliser pendant très longtemps (et encore aujourd'hui) un seuil de 50 000 dollars par année de vie gagnée (les QALY n'existaient pas à l'époque), tout du moins aux États-Unis.
L'OMS a établi une référence fondée sur le PIB par tête, en recommandant une fourchette plutôt qu'un seuil : tout investissement en santé dont le RDCR est inférieur ou égal au PIB par tête est raisonnable. Tout investissement dont le RDCR serait compris entre une fois le PIB par tête et trois fois le PIB par tête doit être soigneusement étudié et choisi en fonction de critères supplémentaires. Au-delà, il ne serait pas recommandé. Cette recommandation repose donc essentiellement sur la capacité à payer d'un pays et dans ce sens elle est facilement compréhensible.
Au Royaume-Uni, les seuils mis à jour en 2014 sont de 20 000 livres sterling (environ 23 000 euros) et de 50 000 livres sterling (environ 58 000 euros, taux de change de septembre 2021). À cette fourchette sont associées des recommandations en termes d'accès au remboursement ; en dessous du premier seuil, les produits seront recommandés ; entre les deux seuils, ils ne seront recommandés que sur la base d'arguments supplémentaires : le fardeau de la maladie, son impact sociétal, le degré d'incertitude sur le RDCR, la difficulté de mesurer la qualité de vie, le caractère innovant du traitement et la prise en compte de dimensions au-delà de l'amélioration de la santé par le NHS (par exemple, d'équité dans l'accès aux innovations). Au-delà du deuxième seuil, le NICE recommande de ne pas prendre en charge les produits (Devlin et Parkin, 2004).
La métrique choisie est celle des QALY, déjà évoquée plus haut. Prenons l'exemple du psoriasis, en présentant un modèle fictif à titre pédagogique. Le bénéfice pour un patient d'un traitement efficace, c'est-à-dire qui supprime de façon durable les éruptions cutanées provoquées par la maladie, est de l'ordre de l'amélioration de sa qualité de vie : disparition des symptômes associés aux plaques, de la gêne ressentie liée à la perception de l'atteinte de son image, etc. Plus cette rémission sera longue, plus ce bénéfice sera important. C'est ce que les économistes ont formalisé avec la métrique du QALY. Un traitement sera donc d'autant plus valorisé qu'il a un impact important et durable sur la qualité de vie du patient relative à son état de santé (health related quality of life, HRQoL).
La métrique utilisée est simple dans son principe : si on est capable de mesurer la qualité de vie liée à un état de santé sur un score qui va de 0 (la mort) à 1 (la parfaite santé), alors le bénéfice d'un traitement par rapport à un autre est égal au différentiel de durée de vie qu'il permet multiplié par le différentiel de qualité de vie qu'il produit. Dans notre exemple hypothétique et simplifié du psoriasis, si on sait que l'indice de qualité de vie d'un patient avec des plaques persistantes est de 0,7 et qu'il subit cette gêne pendant six mois par an, alors durant cette année, il va cumuler six mois avec une qualité de vie parfaite valant un et six mois avec une qualité de vie dégradée de 0,7. Au total, cette année « vaudra » pour lui 0,5 × 1 + 0,5 × 0,7, soit 0,85. Si un nouveau traitement permet de réduire la durée de la poussée de moitié, de six mois à trois mois, le même calcul conduit à une « valeur » de : 0,75 × 1 + 0,25 × 0,7 = 0,925. Le nouveau traitement lui apportera donc un bénéfice de 0,925 – 0,85 = 0,075 QALY sur l'année.
Pour réaliser ces calculs, ce dont l'économiste a besoin est un instrument générique décrivant des états de santé qui se différencient les uns des autres par une plus ou moins bonne qualité de vie et qui peuvent s'appliquer à plusieurs maladies. Autrement dit, cet instrument de mesure de la qualité de vie aura la prétention de s'appliquer aussi bien à un patient cancéreux qu'à un patient souffrant de psoriasis, même si à l'évidence les états de santé dans le premier cas seront plus détériorés. À chacun de ces états de santé, l'économiste associera une valeur, attribuée par des techniques d'enquête auprès d'un échantillon représentatif de la population nationale.
Au niveau international, un instrument s'est imposé, sans nul doute grâce à sa simplicité d'administration. Le questionnaire EUROQOL-5D (EQ-5D), développé par un consortium européen de chercheurs, privilégie cinq dimensions de la qualité de vie liée à la santé : la mobilité, l'autonomie, l'impact sur les activités courantes (loisir, emploi, vie sociale), la douleur ou la gêne, enfin le degré d'anxiété ou de dépression créé par la maladie. Chacune de ces dimensions est mesurée sur cinq niveaux. Chaque état de santé est donc une combinaison de cinq valeurs différentes. Une enquête en population générale permet d'attribuer à chaque état de santé ainsi défini une valeur comprise entre 0 (la mort) et 1 (la parfaite santé). Un autre terme est utilisé couramment pour définir ces valeurs : c'est celui d'utilité (utility). Ces valeurs ont été calculées en France (Andrade et al., 2020).
On pourrait donc présenter le modèle anglais comme un cas où les entreprises ont la liberté de fixer leur prix, mais où elles n'ont pas la certitude de pouvoir vendre à ce prix. La réalité est plus complexe : le modèle de décision a conduit à rejeter la prise en charge de traitements nouveaux en cancérologie. Ces décisions ont été suffisamment impopulaires pour que le gouvernement Tory de John Major crée un fond spécial cancer échappant à la logique du seuil. Dans un deuxième temps et de façon plus pragmatique, le gouvernement anglais a ouvert les négociations sur les prix. Par le biais de programmes qui s'intitulent « Patients Access Schemes », le NHS se donne la possibilité d'obtenir des remises permettant aux traitements de passer sous la barre des seuils. Autrement dit, ce qui fut autrefois l'un des archétypes de marché à prix libres est maintenant passé aux marchés négociés.
Le modèle français saisi par l'utilitarisme ?
La cohérence forte du modèle anglais, par rapport au modèle français qui évoque des négociations secrètes fondées sur des rapports de force et des compromis, a conduit en 2013 à introduire en France l'obligation pour les laboratoires revendiquant un ASMR I, II ou III et donc un prix libre, de présenter des études coût-efficacité sur des bases très proches du modèle anglais. C'est la CEESP déjà évoquée qui a la responsabilité d'évaluer les études présentées par les entreprises, en même temps que la CT procède à une évaluation médicale et de santé publique. Le rapport de la CEESP sur le dossier économique, dit avis d'efficience, est transmis en même temps que les rapports des autres commissions au Comité économique des produits de santé, le CEPS, avec lequel la négociation de prix va s'engager. Mais à la différence de l'Angleterre, la valeur du ratio coût/efficacité ne conditionne pas l'accès au remboursement. Il n'y a d'ailleurs pas de seuil fixé en France. Les résultats des études sont censés aider le CEPS dans ses négociations de prix, mais cet impact est pour le moment de second ordre. En effet, si le produit voit son ASMR I, II ou III confirmé, il a la garantie d'obtenir le prix qu'il a demandé. S'il obtient un ASMR IV ou V alors qu'il a demandé plus, il rentre dans le processus classique de négociation. Les résultats de ces études ont un impact seulement dans le cas où la CEESP estime que soit les données existantes ne permettent pas réellement d'établir l'efficience du produit, malgré la qualité de l'étude réalisée, soit que l'industriel n'a pas respecté le cahier des charges établi par la HAS. Dans ces cas, le CEPS peut s'affranchir de la clause du prix européen.
Est-il envisageable que cette situation change ? À défaut que l'on choisisse un seuil en France, ce qui serait contraire aux fondamentaux de notre système de protection sociale, les résultats des études permettraient, au fur et à mesure que leur nombre augmenterait, de créer un référentiel de ratio et donc de prix, par exemple par classe thérapeutique ou par niveau d'ASMR. Cela donnerait alors au payeur un autre ancrage pour fixer son objectif de prix.
Conclusion
On le voit, la fixation du prix des médicaments est le résultat d'un rapport de force entre l'industrie et les payeurs. Dans cette confrontation, le payeur se trouve dans une situation forte d'asymétrie d'information sur la logique de formation des prix par les entreprises, situation d'autant plus inconfortable que les produits apportent des promesses majeures d'amélioration des traitements. En contrepartie, en France comme au Royaume-Uni, le payeur est en position de monopsone. Cet exposé s'est limité aux « idéaux types » principaux du marché européen. À titre informatif, le gouvernement japonais se caractérise par une régulation encore plus administrée des prix que ce que l'on vient de présenter. Seul le marché nord-américain peut être considéré comme un marché de prix négociés entre chaque entreprise et les géants de l'assurance en santé. Dans le contexte actuel d'une compétition internationale autour de l'innovation en santé, la position du payeur est compliquée par la prise en compte de l'objectif de créer un contexte favorable et attractif pour les implantations de recherche et de production et de soutien aux start-up nationales.
Prix administrés ou prix négociés ? L'un et l'autre à la fois : on a coutume de parler en France d'une politique conventionnelle, suggérant que les deux parties préfèrent une logique contractuelle d'engagements réciproques. En tout état de cause, la négociation se fait dans un cadre confidentiel de protection du secret des affaires, ce qui entretient un soupçon sur une collusion éventuelle entre les entreprises et les payeurs, alors même qu'en France, du moins, la maîtrise des prix des médicaments a été extrêmement efficace au cours des trente dernières années (de Pouvourville, 2021).